Mapping and Variant Calling¶
In this practical you will learn to map NGS reads to a reference sequence, check the output using a viewer software and investigate some aspects of the results. You will be using the read data from the Quality Control practical.
EHEC O157 strains generally carry a large virulence plasmid, pO157. Plasmids are circular genetic elements that many bacteria carry in addition to their chromosomes. This particular plasmid encodes a number of proteins which are known or suspected to be involved in the ability to cause severe disease in infected humans. Your task in this practical is to map your prepared read set to a reference sequence of the virulence plasmid, to determine if the pO157 plasmid is present in the St. Louis outbreak strain.
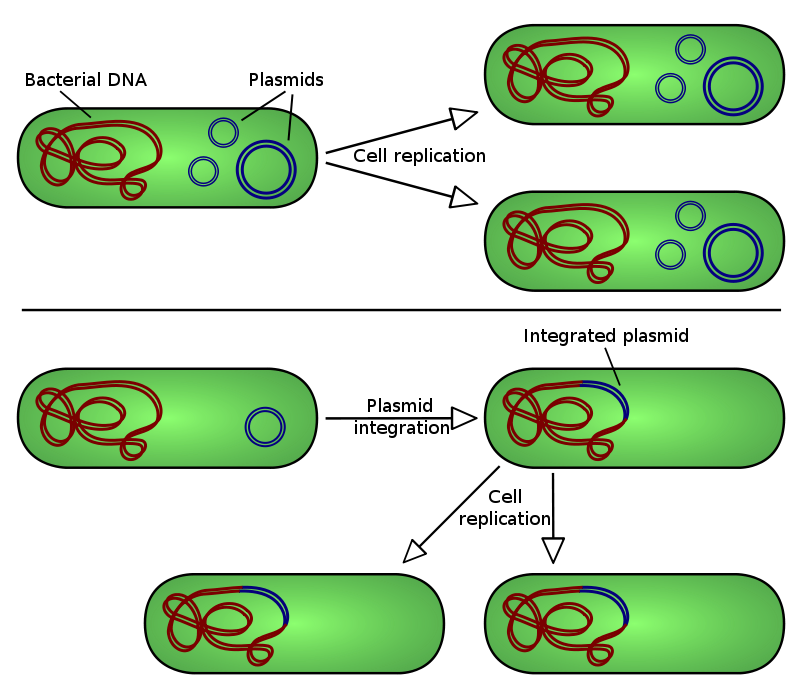 Illustration of plasmid integration into a host bacteria
Illustration of plasmid integration into a host bacteria
Downloading a Reference¶
You will need a reference sequence to map your reads to.
cd ~/work
curl -O -J -L https://osf.io/rnzbe/download
This file contains the sequence of the pO157 plasmid from the Sakai outbreak strain of E. coli O157. In contrast to the strain we are working on, this strain is available as a finished genome, i.e. the whole sequence of both the single chromosome and the large virulence plasmid are known.
Indexing the reference¶
Before aligning the reads against a reference, it is necessary to build an index of that reference
bowtie2-build pO157_Sakai.fasta.gz pO157_Sakai
Note
Indexing the reference is a necessary pre-processing step that makes searching for patterns much much faster. Many popular aligners such as Bowtie and BWA use an algorithm called the Burrows–Wheeler transform to build the index.
Aligning reads¶
Now we are ready to map our reads
bowtie2 -x pO157_Sakai -1 SRR957824_trimmed_R1.fastq.gz \
-2 SRR957824_trimmed_R2.fastq.gz -S SRR957824.sam
The output of the mapping will be in the SAM format. You can find a brief explanation of the SAM format here
Note
In this tutorial as well as many other places, you'll often see the terms mapping and alignment being used interchangeably. If you want to read more about the difference between the two, I invite you to read this excellent Biostars discussion
Visualising with tview¶
head SRR957824.sam
But it is not very informative. We'll use samtools to visualise our data
Before downloading the data in tablet, we have to convert our SAM file into BAM, a compressed version of SAM that can be indexed.
samtools view -hSbo SRR957824.bam SRR957824.sam
Sort the bam file per position in the genome and index it
samtools sort SRR957824.bam SRR2584857.sorted.bam samtools index SRR2584857.sorted.bam
Finally we can visualise with samtools tview
samtools tview SRR2584857.sorted.bam pO157_Sakai.fasta.gz
Tip
navigate in tview:
- left and right arrows scroll
- q to quit
- CTRL-h and CTRL-l scrolls more
- g gi|10955266|ref|NC_002128.1|:8000 will take you to a specific location.
Variant Calling¶
A frequent application for mapping reads is variant calling, i.e. finding positions where the reads are systematically different from the reference genome. Single nucleotide polymorphism (SNP)-based typing is particularly popular and used for a broad range of applications. For an EHEC O157 outbreak you could use it to identify the source, for instance.
We can call the variants using samtools mpileup
samtools mpileup -uD -f pO157_Sakai.fasta.gz SRR2584857.sorted.bam | \ bcftools view - > variants.vcf
You can read about the structure of vcf files here. The documentation is quite painful to read and take a look at the file
Look at the non-commented lines
grep -v ^## variants.vcf
The first five columns are CHROM POS ID REF ALT.
Use
grep -v ^## variants.vcf | less -S
for a better view.
Tip
Use your left and right arrows to scroll horizontally, and q to quit.
Question
How many SNPs did the variant caller find? Did you find any indels?
Examine one of the variants with tview
samtools tview SRR2584857.sorted.bam pO157_Sakai.fasta.gz \ -p 'gi|10955266|ref|NC_002128.1|:43071'
That seems very real!
Question
Where do reference genomes come from?